







Review - Journal of Molecular Pathophysiology (2023)
Distinguishing Therapeutic Hypoxia from Pathogenic Hypoxia: Benefits for Cardiac Patients
Kieran Oldfield1,2,5, Rohan Jayasinghe2,3,4 and Gillian Renshaw5*2Department of Cardiology, Gold Coast University Hospital, Queensland, Australia
3Department of Medicine, Bond University, Queensland, Australia
4Department of Medicine, Macquarie University, New South Wales, Australia
5Department of Health Sciences and Social Work, Griffith University, Queensland, Australia
Gillian Renshaw, Department of Health Sciences and Social Work, Griffith University, Queensland, Australia, Email: g.renshaw@griffith.edu.au
Received: 17-May-2023, Manuscript No. JMOLPAT-23-98986; Editor assigned: 19-May-2023, Pre QC No. JMOLPAT-23-98986 (PQ); Reviewed: 05-Jun-2023, QC No. JMOLPAT-23-98986; Revised: 12-Jun-2023, Manuscript No. JMOLPAT-23-98986 (R); Published: 19-Jun-2023
Abstract
Therapeutic intermittent hypoxia is a non-pharmacological lab-based intervention with the demonstrated benefits of ascent to moderate altitude. Depending on the dose or severity of therapeutic intermittent hypoxia or high-altitude, hypoxic exposure can stabilise master gene regulators to instigate beneficial adaptations. Therapeutic intermittent hypoxia has been demonstrated to initiate adaptive phenotypic changes such as: decreased sympathetic input with a decrease in systolic blood pressure; increased haemoglobin and red blood cell count; altered substrate metabolism to favour increased glucose uptake and fatty acid metabolism with decreased fatty acid synthesis; increased antioxidant defence; decreased inflammatory cytokines. Such beneficial adaptive changes to therapeutic intermittent hypoxia serve not only to diminish the impact of cardiovascular pathology but also to increase exercise tolerance.
Several lines of evidence indicate that therapeutic intermittent hypoxia may provide a new treatment modality for people affected by heart failure with a reduced ejection fraction. This review aims to identify links in the literature between heart failure pathophysiology and the beneficial adaptations induced by TIH.
Keywords
Heart failure, Intermittenthypoxia, Phenotypic adaptations, Haemoglobin, Red blood cells, Sympathetic input.
Introduction
Epidemiological data demonstrates that Heart Failure (HF) is increasingly prevalent which places a significant cost upon society and the individual. The estimated 5-year all-cause mortality from HF with reduced ejection fraction (HFrEF) ranges from 56% to 75% [1,2] and has an estimated global cost of US $ 108 billion [2]. The cause of death for more than half of HFrEF population was “cardiovascular death” [1]. HF can result from multiple medical conditions including Myocardial Infarction (MI), Ischaemic Heart Disease (IHD), and Hypertension (HTN) [3]. In Western countries there is an estimated prevalence of 11% for all HF [4]. It can be separated into two separate types with different pathophysiologies: HF with a Reduced Ejection Fraction (HFrEF) or with a preserved ejection fraction (HFpEF). Heart failure with a reduced ejection fraction is characterised by impaired contraction of the left ventricle (systolic dysfunction) and reduced Left Ventricular Ejection Fraction (LVEF) with an estimated prevalence of 5.5%. Medical management of HF has evolved over the last few decades with the introduction of pharmaceuticals to treat high blood pressure, Beta Blockers (BB), Angiotensin- Converting-Enzyme (ACE) inhibitors, aldosterone antagonists and diuretics to diminish fluid retention. Neprilysin inhibitors (ARNI) have been developed which not only treat high blood pressure, such as ACE inhibitors, but also induces vascular dilation and prolongs the effect of Atrial Naturetic Peptide (ANP) and Brain Derived Naturetic Peptide (BNP), which assist in compensating for HF. Recently inhibitors of Sodium-Glucose Cotransporter 2 (SGLT2) have been introduced as one of the “four pillars” of pharmacotherapy for HFrEF [5,6]. With the introduction of each of these agents we have seen a reduction in mortality and morbidity in HF patients [7]. Despite these advances there is still ongoing high morbidity and mortality for HF patients.
Nonpharmacological interventions such as cardiac rehabilitation, alterations to diet and regular moderate exercise have been shown to be beneficial adjuncts to medical therapy in decreasing morbidity for HF [8, 9]. There is growing interest in using novel interventions such as doses of Intermittent Hypoxia (IH) as a therapy (TIH) to provoke beneficial adaptions in cardiovascular and metabolic diseases. In adult humans, beneficial phenotypic responses to laboratory based TIH, at a fixed SpO2, has resulted in diminished key cardio-metabolic risk factors, demonstrating the therapeutic potential of a specific hypoxic pattern as non-pharmacological intervention [10]. In healthy seniors TIH resulted in increased haematocrit and red blood cell number [11]. In addition, exposure to a fixed fraction of inspired oxygen (FiO2) prior to an altitude challenge induced anti-inflammatory pathways, diminished reactive oxygen species production, increased antioxidant defence, and unregulated fatty acid metabolism which serve to restrict hypoxia-induced inflammation, dyslipidemia as well as regulating redox homeostasis [12]. After 10 sessions of TIH, in a healthy adult population, significant beneficial changes to metabolism occurred: (i) average arterial glucose was diminished by 0.9 mmol L-1 (ii) average Low-Density Lipoprotein (LDL) was diminished by 0.5 mmol L-1 resulting in an improved LDL/HDL ratio; and (iii) systolic blood pressure was decreased [10]. On a fundamental level, there is clear evidence that exposure to hypoxia activates Hypoxia Inducible Factor 1 alpha (HIF-1α), a transcription factor regulating the expression of over 1000 genes including an increase in the levels of erythropoietin (EPO) and Vascular Endothelial Growth Factor (VEGF) [13]. The expression of these oxygen-sensitive genes not only results in an elevated red blood cell number but also in an increased vascular network [12,13], both of these hypoxia-induced adaptations are expected to benefit HF patients with anaemia.
Literature Review
Discriminating between involuntary breath-holding and therapeutic intermittent hypoxia
Previously, the safety and utility of TIH was questioned because it was assumed to have similar effects to the Intermittent Hypoxia (IH) experienced during involuntary breath-holding in either Obstructive Sleep Apnoea (OSA) or Central Sleep Apnoea (CSA), both of which involve breath holding and hypercapnia. In contrast to sleep apnoea, neither IH nor TIH have a hypercapnic component. Multiple lines of evidence demonstrate that OSA and CSA produce maladaptive systemic responses such as hypertension and insulin resistance [14,15]. Emerging research has demonstrated that beneficial adaptive changes can occur in response to certain types of hypoxic challenge, depending on the dose, duration, or pattern of hypoxia/reoxygenation protocols [11,14]. Therapeutic intermittent hypoxia at a fixed SpO2 of 85% did not elevate the level of cortisol, secretory immunoglobulin A, or cardiac troponin T demonstrating the safety of TIH in healthy seniors [11].
A diminished supply of oxygen can result in either adaptive and maladaptive change in two distinct states: During hypoxaemia induced by OSA and CSA (representing involuntary breath-holding), where there is a lowered partial pressure of oxygen in the blood; while during hypoxia which occurs without breath holding, there is a diminished oxygenation of tissues [16]. The maladaptive outcomes of OSA impairs metabolism as well as having a negative impact on cardiac, cognitive, and respiratory function [17]. In contrast, the adaptive outcomes of exposure to hypoxia include diminished cardiovascular risk factors and hypertension [10,14].
However not only do tissues differ in their sensitivity to hypoxia due to their individual metabolic requirements [18] but also individuals desaturate to a different extent in response to the same FiO2. Successful application of TIH is linked with (i) a standardised target oxygen saturation in the venous (SpO2) or arterial (SaO2) circulation by altering FiO2; and (ii) the same duration of hypoxic exposure, thus providing an equivalent hypoxic dose for all participants. More specifically, the dose consists of exposure to the same number of total minutes of hypoxia (in dispersed with brief periods of normoxia) while maintaining the same mean target for SpO2 [11].
In contrast, to OSA and CSA, TIH does not have a hypercapnic component, nor does it elevate Sympathetic Nerve Activity (SNA) [19]. These differences distinguish hypoxia generated by involuntary breath-holding in OSA/CSA from laboratory-based hypoxia administered in TIH. In addition, TIH lowers cardiovascular risk factors by: (i) decreasing fasting glucose levels, with changes persisting for up to a month post-cessation of TIH [20]; (ii) lowers circulating lipid levels, particularly LDL [10,14]; and (iii) decreased insulin resistance [21].
While it is acknowledged that ongoing inflammation results in cardiac dysfunction, the causal relationship is not entirely clear, nor is it clear whether the effect of inflammation is similar across different heart failure phenotypes [22]. Patients treated with neurohormonal antagonists, specifically angiotensin-converting enzyme inhibitors and β-adrenergic receptor blocking agents have been shown to have a lower amount of circulating pro-inflammatory cytokines, suggesting a causal relationship between inflammation and cardiac dysfunction [22]. It has been postulated that ongoing inflammation within malfunctioning tissues is a para-inflammation, i.e. inflammation that is not a direct result of infection or acute injury. Instead, it is a sustained tissue injury or a failed downregulation of the initial inflammatory response tissue results in an inability to restore homeostasis and on-going inflammation [23].
Comparison of laboratory based hypoxia versus ascent to altitude: differences in hypoxic patterns
Laboratory based IH protocols differ in the FiO2, as well as the number and duration of daily cycles administered to patients [14]. An FiO2 between 9-16% with low cycle numbers (3-15 per day), have been shown to initiate beneficial adaptations. Conversely, severe IH protocols using an FiO2 of 2-8% had pathological effects that were like those reported for OSA/CSA induced IH [14]. From a safety perspective, TIH and resultant elevation in HIF1 α stabilisation in response to an FiO2 of 10-12% has been shown to be safe and to have a beneficial impact on key cardiovascular risk factors such as hypertension, glucose, decreasing sympathetic input via an altered vagal tone, lipid levels and lowers the formation of atherosclerotic plaque [10]. Other effects include diminishing inflammation, body weight, increasing aerobic exercise capacity, and diminishing myocardial injury in ischaemic heart disease as detailed below [10, 12, 13, 24-27].
The key differences between ascent to altitude and lab based IH are twofold: its duration and the pattern of the hypoxic stimulus it provides. Unlike laboratory- based hypoxia, the ascent to altitude has no optional periods of reoxygenation so it provides a continuous stimulus, which increases in intensity with increased altitude. Stabilisation of HIF-1α in response to periods of low oxygen (hypoxia) orchestrates the expression of oxygen sensitive genes that mediate beneficial adaptations to hypoxia [28]. It was hypothesised that the repeated presentation of a hypoxic stimulus, as seen in TIH may be needed to provoke adaptive changes in phenotype, intermittent but not chronic hypoxia, of the same duration, resulted in increased haemopoesis in healthy seniors, like that achieved by acclimatization to altitude [11]. While the mean SpO2 was set to 85% with the same total minutes in hypoxia for both hypoxic patterns, there were two key differences between the intermittent and continuous hypoxic pattern that could potentially affect adaptive phenotypic change: (i) the nadir of SpO2 was lower in intermittent hypoxia to make up for the periods of reoxygenation; and/or (ii) repeated reoxygenation during IH would block the stabilisation of HIF-1α in a cyclic pattern corresponding to desaturation of tissues in the alternating hypoxic intervals.
Benefits of stabilising hypoxia inducible factor 1α, in response to TIH
Exposure to normobaric hypoxic patterns increases the level of HIF-1α, which has been demonstrated to regulate of over 1000 genes, including but are not limited to: (i) EPO, resulting in increased haemopoesis [13]; (ii) VEGF, stimulating capillary growth [13,24,29]; (iii) pathways involved in glucose and lipid metabolism [30,31]; and the activation of a number of molecular switches that initiate beneficial adaptive change, two of which are discussed below. Elevated levels of HIF- 1α shift energy production from oxidative metabolism towards glycolytic metabolism, possibly explaining the lowered serum glucose post-TIH shown in some studies [10,13,32]. Similarly, when the oxygen induced breakdown of HIF-1α and HIF-2α is inhibited, by exposure to hypoxia, a decrease in cholesterol and fatty acid synthesis is observed [25]. In the same study an increase in lipoprotein lipase, and glycolytic enzymes was observed when HIF-1α and HIF-2α breakdown was inhibited [25]. Therapeutic intermittent hypoxia not only increases HIF-1α but also increases nitric oxide metabolites and lowers systolic blood pressure [32] in hypertensive patients.
Diminished cardiovascular risk factors after therapeutic intermittent hypoxia
Elevated glucose and/or lipid levels are risk factors for cardiovascular events, and there is evidence that exposure to several types of TIH can diminish risk factors for cardiovascular events. Treatment with TIH can decrease serum glucose post-IH by increasing skeletal muscle glucose uptake in humans [20,33]. Treatment with moderate regimes of intermittent hypoxia increased glucose uptake in skeletal muscle, via a multifunctional molecular switch, AMP-activated protein kinase (AMPK) activation [34]. In addition, during hypoxia there was an increase in skeletal muscle 3-0-methylglucose transport, and an upregulation in glucose transporter 4 (GLUT-4) within the skeletal muscle plasma membrane, mediated by AMPK [35,36].
There is evidence that exposure to TIH results in lowered circulating lipid levels [10, 21]. This is important because increased LDL levels are associated with increased atherosclerosis [37], a major cardiovascular risk factor. Low density lipoprotein is catabolised by two separate pathways. The receptor-dependent pathway involves LDL binding to apolipoprotein B-100 resulting in hepatic endocytosis and breakdown of the LDL molecule. While the receptor-independent pathway occurs in non-hepatic tissues and increases in parallel with an elevation in LDL [37]. It has been demonstrated that TIH affects lipid levels and has a more pronounced effect on LDL than HDL [10,12]. The lowered LDL levels can be explained in terms of the hypoxia induced stabilization of HIF-1α which lowers cholesterol and fatty acid synthesis and increases lipoprotein lipase [25]. A study using moderate TIH at an FiO2 of 12% for 4 hrs as pre-conditioning, prior to 7 days of hypobaric hypoxia at 3500m (FiO2 of 13.5%) showed a reduction in LDL levels and changes in several proteins involved in relipid regulation [12]. The Apolipoprotein A1 (Apo A1) component of HDL was increased compared to baseline measurements post-IH and on day 7 at 3500m. The levels of apolipoprotein B-100 (Apo B-100), involved in the LDL degradation pathway, increased, or decreased proportionately in relation to LDL levels [12]. The levels of Apo B-100 and LDL were found to decrease in response to TIH and hypobaric hypoxia [12,37]. These tandem changes in HDL and LDL would beneficially lower the LDL to HDL ratio, representing a healthier state. In addition, Paraoxonase 1 (PON1), an enzymatic component of HDL, with significant anti-atherosclerotic properties, was significantly increased post-IH exposure [12]. While elevated HIF-1α shifts energy derivation from oxidative metabolism towards glycolytic metabolism, the concomitant increase in AMPK also stimulates glycolytic enzymes, which may contribute to the lowered serum glucose level observed in hypoxia studies [13,36].
The metabolite AMPK is activated through multiple pathways, one of the most recognised being hypoxia [36]. It appears to be integral to glucose metabolism and deficiencies in AMPK are associated with arrythmias and hypertrophic cardiomyopathies [36]. Insulin levels have been found to either increase or decrease in response to hypobaric hypoxia [38,39]. It is unclear whether insulin is responsible for the observed reduction in serum glucose observed during hypobaric hypoxia and TIH. in addition, TIH has been shown to result in a reduction in both fasting glucose levels and post prandial glucose levels [21]. Type 2 diabetes mellitus (T2DM) and glucose variability are both known risk factors for the development of coronary artery disease [40,41], as such TIH could be a potential treatment modality to diminish cardiovascular risk factors.
The elevation of AMPK levels in response to exposure to high altitudes [12,42] and acts as a molecular switch in key pathways including but not restricted to (i) endothelial cell function; (ii) non-NOS vasodilation; (iii) the metabolism of glucose; (iv) the metabolism of lipids; sensing cellular energy; (vi) redox sensor; and (vii) mitochondrial quality control.
The vasodilation of arteries, regulation of inflammation and proliferation of Vascular Smooth Muscle (VSM) was demonstrated to be governed by AMPK, [36]. Activation of AMPK relaxes VSM by lowering Ca2+ and by reducing the sensitivity of the arterial contractile machinery to Ca2+ [36]. These two AMPK governed mechanisms are independent of exogenous Nitric Oxide (NO) or the NO produced by the vascular endothelium (eNOS) [36]. Activation of AMPK protects against vascular calcification and exerts antimigratory and antiproliferative actions in vascular smooth muscle cells [36]. In addition, AMPK activation directly activates endothelial nitric oxide synthase (eNOS) increasing the production of Nitric Oxide (NO) production and its bioavailability [43]. Nitric oxide is a crucial to normal endothelial function unless its bioavailability is diminished by inflammation, oxidative stress or lipid infiltration [43]. The diminished bioavailability of NO and subsequent impaired vasoconstriction is an early stage of atherosclerosis. Furthermore, AMPK is crucial for normal endothelial function and is protective against atherosclerosis and arterial calcification [36,43].
The formation of AMPK is instrumental in the regulation of fatty acid uptake and metabolism with increased activity resulting in decreased fatty acid synthesis and the increased oxidation of fatty acids [44] via phosphorylation. More specifically, the oxidation of fatty acids and their biosynthesis are regulated by two derivatives of acetyl coenzyme A carboxylase (alpha ACC1 and beta ACC2) which are both phosphorylated by AMPK which decreased fatty acid synthesis as well as fatty acid oxidation, with the subsequent improvement in lipid profile [44]. The activation AMPK inhibits the synthesis of TAGs resulting in a reduction in lipid storage. Finally, AMPK increases the catabolism of fatty acids, and promotes intracellular transport of fatty acids to allow greater mitochondrial oxidation [36]. As such hypoxia induced AMPK levels result in lipid reduction and a protective state against metabolic syndrome [42,44]. Taken together these AMPK regulated mechanism may explain why TIH decreased LDL and an increased HDL concentrations [10]. In conjunction with lowering the level of LDL, a risk factor for atherosclerosis, HIF-1α lowers the formation of atherosclerotic plaque in animal models [12,13,24-27].
Evidence has confirmed that AMPK acts as a sensor of cellular energy that can be activated through diverse pathways, one of which is in response to hypoxia [36,43,45]. While it is known that hypoxic exposure increases AMPK activation [34], HIF-1α has been posited to regulate AMPK activation, via the coupling of HIF-prolyl-4-hydroxylases with Ca2+, but this is controversial [45]. Elevation in AMPK levels has discreet actions upon metabolic regulation and ultimately health. Lipid and glucose metabolism and homeostasis are discussed above. Other areas that are affected by elevated AMPK include, inflammatory regulation, mitochondrial function, vascular and endothelial function, and it has been shown to exert protective effects on various conditions including cardiomyopathies and metabolic syndromes [36,43,44].
Reactive Oxygen Species (ROS) are produced as a by-product of normal cell function [36]. Hyperlipidaemia, insulin resistance, obesity, inflammation, and hyperglycaemia all cause abnormally elevated levels of ROS and subsequent oxidative stress [36]. As a relipid sponse to many pro-atherosclerotic stimuli, mitochondria become dysfunctional, leading to a direct increase in mitochondrial ROS generation [36,43]. Increased oxidative stress and inflammation result in endothelial dysfunction, which is considered a critical precursor to atherosclerosis via numerous mechanisms [43]. The metabolite AMPK acts as a redox sensor in a feedback loop, in which increased levels of ROS directly activate AMPK [45]. Once activated AMPK maintains redox status by inhibiting the production of oxidants, modulating antioxidant gene expression increasing the cellular antioxidant potential, and by regulating mitochondrial homeostasis [36,43]. Over expression of AMPK or the in vivo activation of it has been demonstrated to normalise endothelial function in diabetic rats. Upregulation of AMPK also mitigates superoxide generation through the uncoupling of Endothelial Nitric Oxide Synthase (eNOS) [36]. However, AMPK can become disordered in patients with hypertension or T2DM, diminishing its capabilities to normalise endothelial dysfunction as well as inflammation [36,46].
Signalling via AMPK also plays an important role in response to changes in energy expenditure or cellular energy stress as well as its role in the quality control of mitochondria. Intracellular mitochondrial are regulated via biogenesis, to either form new mitochondria or to remove damaged mitochondria via autophagy/ apoptosis to remove damaged mitochondria [36,43]. In addition, mitochondrial metabolism is predominantly regulated by AMPK acting in conjunction with transcription factors from the peroxisome proliferator- activated receptor gamma coactivator (PGC) family, [43,46], which act as molecular switches not only to alter mitochondria plasticity but also their fate. Mitochondrial biogenesis is also regulated by the interaction between AMPK and PGC1α to increase mitochondrial number in response to demand [43]. Conversely, during cellular energy stress AMPK activates the unc- 51-like autophagy activating kinase 1 (UKL1) directly and indirectly through the inhibition of mammalian target of rapamycin complex 1 (mTORC) resulting in the removal of damaged mitochondria and cellular components via autophagy, providing an important component of mitochondrial quality control [43,46]. In summary, AMPK activation effectively maintains cellular and mitochondrial homeostasis.
Ischaemic preconditioning
Another promising therapeutic application of TIH is in preconditioning the human body to withstand a subsequent hypoxic event, termed Ischaemic Preconditioning (IPC). Whole body ischemic and hypoxic conditioning can have beneficial protective effects on multiple organ systems even while temporarily stopping blood flow to a specific organ [47]. Myocardial Infarcts (MI) are reduced in size when exposed to IPC [13,24,25]. In animal models, exposure to localised ischaemia results in collateral blood flow and vascular remodelling via induced IPC, it lowers the extent of damage from ischaemic insults [13, 27]. The induction of HIF-1α appears to be integral to the process of ICP as stabilisation of HIF-1α results in smaller myocardial infarcts [26]. Animal models show that complete knockout of HIF-1α (HiF-1α−/−) is not compatible with life. A partial knockout of HIF-1α+/− were viable [13]. The vascular changes from ICP did not occur in HIF-1α partial knockouts showing HIF-1α is necessary for ICP. HIF-1α stabilisation in heart failure patients results in the activation of VEGF to promote neovascularisation [13]. Preconditioning with TIH in animal models led to a reduction in the size of myocardial infarctions occurring in response to ischemia-reperfusion injury (I-R) [13, 24, 27].
While multiple factors are involved in protection from IPC, including the production of local hormones (autacoids), HIF-1α is strongly implicated as the master regulator of adaptive changes in phenotype in response to oxygen limitation [13,24,48]. In fact, hypoxia-induced stabilisation of HIF-1α has been shown to significantly diminish I-R injuries [26]. The duration of the time interval between hypoxic preconditioning and conferred protection varied between studies, with one study showing a benefit at 24 hrs post-preconditioning but not at 30 minutes [24]. Other sources suggest that both an immediate and a delayed phase of hypoxia-induced protection occurs against subsequent ischaemic injuries [13]. There is an immediate form of IPC that lasts for up to 2-3 hours following a post-hypoxic stimulus. This immediate effect subsides and is then followed by a second delayed protective effect occurring within 12- 24 hours and may last up to 72 hours. These two protective phases of IPC are thought to represent different signalling mechanisms [48]. The evidence and underlying molecular mechanisms involved in cardio-protection by IPC has been extensively reviewed by Mallet and colleagues [49] and since it involves a cycle of hypoxia followed by reoxygenation, it shares common pathways with TIH.
To assess the role of HIF in cardiac protection: mice with a heterozygous knockout allele at the HIF locus, were exposed to IPC prior to an induced MI. This subgroup of mice experienced no protective effects of ICP and had similar infarct sizes and decline of cardiac function as mice that were not preconditioned with IPC [24], providing evidence that a HIF1 α cascade is needed to provide protection from an induced MI. Interestingly, mice pre-treated with an infusion of EPO, then subjected to I-R had a preservation of myocardium compared to controls [24], confirming that one of the key signals triggered by HIF1 α, prevented ischemia reperfusion injury. Ischaemic preconditioning does not require a local stimulus to provide an adaptive advantage. This phenomenon labelled “remote ischaemic conditioning” was initially identified when a hypoxic stimulus is applied to a single coronary artery resulted in protection in other coronary arteries [50]. Remote ischaemic conditioning has further been shown to be protective when the ischaemic stimuli is not provided to the target organ i.e. hypoxia of a limb or kidneys provide an adaptive advantage to the heart [48]. Remote ischaemic conditioning appears to be reliant on neurohormonal pathways to provide a systemic benefit [48].
Discussion
A role for TIH in coronary artery disease
The development of coronary artery disease is multifactorial, with the major risk factors being uncontrolled hypertension, hyperlipidaemia, diabetes, smoking, physical inactivity, and advancing age [51]. The significant contribution our society’s disease burden, the primary, secondary, and tertiary prevention of Ischemic Heart Disease (IHD) is an area of intense research. Management of established IHD is directed towards managing cardiovascular risk factors through lifestyle modification and directed medical therapy with the goal of bringing IHD risk factors back into the normal range [51].
Inflammation and increased oxidative stress are recognised as major contributors to the development of IHD [52,53]. Hypoxia, at finely tuned doses, can turn down the expression of inflammatory genes making TIH an ideal candidate for a novel approach to therapeutic intervention [54]. It has long been known that patients with chronic proinflammatory diseases such as rheumatoid arthritis (RA), Systemic Lupus Erythematosus (SLE) and psoriasis all have increased cardiovascular risk compared to the normal population. Moreover, when patients with RA are treated with anti-inflammatory agents, e.g., monoclonal antibodies targeting Tumour Necrosis Factor Alpha (TNF α), they experience a reduction in their vascular event rates [53]. In a similar fashion, the antioxidant enzyme glutathione perioxidase-1 has been shown to have a positive correlation with increased cardiovascular eventfree survival [52]. Immune modulation to treat IHD is not without risk though. Colchicine, a medication long used to suppress inflammation through tubulin suppression and a resultant down regulation of multiple inflammatory pathways has recently been investigated as an adjunct treatment for acute IHD [55,56]. Initial research showed promising results for reduced cardiovascular events, however subsequent investigations revealed that this came at the price of an increased all cause mortality, showing that a nuanced and targeted approach to tackling inflammation in cardiovascular disease is required [55,57,58]. Targeted reduction in inflammatory pathways in heart failure has been reviewed elsewhere [22]. Recognition of inflammation’s causal role in the pathogenesis of atherosclerosis and IHD, has led to an increasing body of literature investigating various anti-inflammatory agents and their impact on atherosclerosis [59].
Patients with ischaemic or non-ischaemic heart failure have been shown to be in a state of sustained myocardial inflammation [22]. One clinical trial identified an elevated C-reactive Protein (CRP) in 57% of all patients with HFrEF [60]. Similarly pro-inflammatory cytokines and chemokines can be identified in the hearts of individuals with cardiomyopathies, but not in normal hearts [22]. The exact mechanism of persistent inflammation in HFrEF is not entirely clear, nor is it clear whether it is causal or the consequence of HF; especially considering many risk factors for HFrEF, including diabetes, HTN and CKD are inherently proinflammatory [22,60]. Neurohormonal activation and haemodynamic overload are implicated as contributing to the proinflammatory state, as resolution of these states through medical intervention results in a lowering of inflammatory markers [22]. The level of inflammation appears to be important to heart function. The pro-inflammatory cytokines TNF α, interleukin 6 (IL-6) and interleukin 18 (IL-18) have been shown to weaken the force of cardiac contraction [61]. Targeted inhibition of specific proinflammatory cytokines have shown mixed results [60,61]. While the role of inflammatory markers in the response to myocardial injury are complex and not fully understood, it has been proposed that various components of the inflammatory response have dual functionality, both in promoting and regulating inflammation depending on the receptor activation [62]. This may explain why inhibition of specific cytokines show mixed results. Hearts with histological evidence of inflammation, that when treated with prednisolone, do experience an increase in their LVEF, suggesting systemic reduction in inflammation is beneficial in HFrEF [22]. Since inflammation is a contributing factor to poor LV function and outcomes in patients with IHD and HFrEF, it is important to assess any impact that TIH may have upon inflammatory markers as well as markers of oxidative stress. Recent studies have focused on TIH which uses evidence based hypoxic protocols in clinical settings, as a therapeutic modality for patients [10,11,63]. Conversely, laboratory- based continuous hypoxia (FiO2 <10%), can induce elevated inflammatory markers [14], increased inflammatory markers also result from a transient sojourn at high altitudes (>3000m above sea level) [64], while intermittent hypoxia with a modest FiO2 decreased inflammatory markers and has numerous therapeutic effects on metabolism and haemodynamics, as discussed above [10,11,64]. Furthermore, exposure to continuous hypobaric hypoxia induced by prolonged sojourns at extremely high altitudes, can result in the elevation of circulating proinflammatory cytokines leading to altitude sickness with vascular leakage and oedema [65]. Ascent to 3000m and 4000m above sea level are required for the severe manifestations of pulmonary and cerebral oedema respectively, while early signs such as headaches and nausea can occur between 2500-3000m [66]. A study designed to assess the utility of preconditioning athletes with IH prior to high altitude training found a transient increase in acute-phase reactants including CRP, serum amyloid A-1 and alpha-1-acidglycoprotein 2 [64]. Conversely, athletes who were preconditioned with IH prior to traveling to altitudes, experienced a more rapid decline in their inflammatory markers [64]. At sea level, an overall shift in circulatory from pro-inflammatory to anti-inflammatory cytokine was achieved in participants who exercised during normobaric hypoxia (FiO2 of 13.5%) compared to controls that exercised in room air (FiO2 of 21%) [67].
Potential adaptive advantage of TIH in heart failure patients
Within the heart failure population approximately 50% of hospitalised patients and 30% of stable patients have anaemia (hemoglobin <13 g/dL in men and <12 g/dL in women) [68]. Anaemia in HF is independently associated with increased hospitalisations and mortality [68]. Anaemia in HF appears to be multifactorial, with true iron deficiency and functional iron deficiency appear to play a large role. Patients without objective anaemia appear to have lower haemoglobin concentrations when compared to control groups, and up to 30% may have underlying iron deficiency, which is a predictor of reduced survival [69]. Even among iron replete patients with heart failure, worsening classification in the New York Heart Association (NYHA) and exercise tolerance have been associated with a reduction in circulating ferritin levels [69]. Amongst anaemic HF patient’s, the predictors of having a lower haemoglobin included, elevated ESR, female gender and low transferrin saturations. As functional class worsens the prevalence of iron deficiency amongst this population increases. NYHA class III and class IV patients have iron deficiency rates of 72% and 100% respectively with anaemia of chronic disease being the most common cause [69]. It is unclear if iron deficiency and anaemia are a cause of or a result of worsening NYHA functional class and disease status, an area of intense ongoing research. Treatment with TIH has been shown to elevate red blood cell numbers and elevate haemoglobin levels in healthy seniors [11], either TIH may have elevated EPO directly via stabilisation of the HIF1 α pathway or it may have provided sufficient stress to turn on a non- EPO dependent pathway, the stress-erythropoiesis pathway described by Kim and colleagues [70].
Raised inflammatory markers were associated with both true iron deficiency and functional iron deficiency in chronic HF patients [69]. Heart failure patients often present with chronic inflammation, cytokines such as TNF α and IL-1β in conjunction with chemokines can be isolated in HFrEF patients but are rarely found in non-failing hearts [22]. Similarly, T lymphocytes and NK cells are observed in histological specimens from HFrEF patients in the absence of any alternative cause [22]. Typically, HF is associated with an increase in CRP and multiple pro-inflammatory cytokines including TNF α and IL-6 [68]. Other effects of increased TNF α and interleukin- 6 that contribute to anaemia are not only an inhibition of EPO production but also the suppression of erythroid progenitor cell proliferation within bone marrow [68]. Furthermore, HF causes diminished renal perfusion and the upregulation of HIF1 α, resulting in an upregulation of EPO. However, the increase in EPO is often lower than expected for the degree of anaemia, suggesting a blunted production of EPO in HF [68] which would maintain the anaemic state. The documented increase in EPO in response to TIH [71]. and the shift in circulating cytokines towards a more anti-inflammatory state [12] suggest that TIH could help ameliorate anaemia in the HF population.
Current therapy for HFrEF involves use of ACEI/angiotensin II receptor blocker, ARNI, BB’s, and mineralocorticoid receptor antagonists, all of which have been shown to independently decrease mortality and increase LVEF [7,72]. Recently SGLT2 inhibitors have also been shown to reduce mortality, morbidity, and hospitalisation [5,6]. TIH-induced increases in LVEF have been documented in rodent models. This increase in LVEF has been demonstrated in both normal mice, as well as mice with heart failure secondary to over-expression of TNF α [73]. The documented increase in EPO [72], the increase in red blood cells in response to TIH [11] is expected to ameliorate or reverse anaemia in the HF population. This is further supported by evidence that TIH significantly increased red blood cell count (7.7% at day 5 of treatment compared to baseline) in healthy seniors compared to their sham controls, without any increase in stress markers such as cardiac troponin T and cortisol [11].
Training under hypoxic conditions has been shown to be beneficial to athletes, improving performance and exercise capacity due to mechanisms explained elsewhere [74]. One of the benefits of TIH is the improvement of non-athletes’ exercise capacity [75].
The effect of TIH on exercise capacity can be assessed
by using functional tests including Cardiopulmonary
Exercise Testing (CPET), or the 6-Minute Walk Test
(6MWT), both of which approximate the peak rate (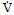 )
of Oxygen Consumption (O2) occurring during exercise
(O2peak). Consequently, measures such as CPET and
the 6MWT reflect O2peak, the individual’s exercise capacity
defined as the body’s ability to take up oxygen
in response to exercise [76]. More specifically, VŻ O2peak is
calculated by the Fick equation, O2 = SV × HR × (CaO2 - CvO2). Where SV is the stroke volume, HR is heart
rate, CaO2 is the arterial oxygen content, and CvO2 is
the mixed venous oxygen content. These components
represent the principal measures of delivery and extraction
of oxygen, [77]. An impairment of any of these
components can diminish O2peak and result in a low O2 [76]. It should be noted that the maximum rate of
oxygen consumption, O2 max is attenuated in HFrEF
patients due to their limited ability to improve their
cardiac output, defined as SV × HR [77,76]. The 6MWT
has been validated as a means of assessing functional
capacity in exercise based cardiac rehabilitation [78].
Evidence suggests that there is an adaptive advantage
in using TIH in cardiac rehabilitation. Therapeutic intermittent
hypoxia has not only been shown to be safe
in individuals with angina and coronary artery disease,
[75] but to also improve both aerobic capacity and exercise
tolerance of participants with and without coronary
artery disease as measured by CPET and the
6MWT [79]. In addition, TIH resulted in an increased
oxygen-carrying capacity which could be linked with
the improvements in fitness [75] which may explain
the reported improvement in fitness measured by
CPET and the 6MWT. Therapeutic intermittent hypoxia
may be particularly useful in HFrEF patients not only
because HFrEF is associated with a reduced exercise
capacity but also because individuals with HFrEF have
a reduced ability to take up oxygen as well as impaired
peripheral oxygen utilisation [77].
)
of Oxygen Consumption (O2) occurring during exercise
(O2peak). Consequently, measures such as CPET and
the 6MWT reflect O2peak, the individual’s exercise capacity
defined as the body’s ability to take up oxygen
in response to exercise [76]. More specifically, VŻ O2peak is
calculated by the Fick equation, O2 = SV × HR × (CaO2 - CvO2). Where SV is the stroke volume, HR is heart
rate, CaO2 is the arterial oxygen content, and CvO2 is
the mixed venous oxygen content. These components
represent the principal measures of delivery and extraction
of oxygen, [77]. An impairment of any of these
components can diminish O2peak and result in a low O2 [76]. It should be noted that the maximum rate of
oxygen consumption, O2 max is attenuated in HFrEF
patients due to their limited ability to improve their
cardiac output, defined as SV × HR [77,76]. The 6MWT
has been validated as a means of assessing functional
capacity in exercise based cardiac rehabilitation [78].
Evidence suggests that there is an adaptive advantage
in using TIH in cardiac rehabilitation. Therapeutic intermittent
hypoxia has not only been shown to be safe
in individuals with angina and coronary artery disease,
[75] but to also improve both aerobic capacity and exercise
tolerance of participants with and without coronary
artery disease as measured by CPET and the
6MWT [79]. In addition, TIH resulted in an increased
oxygen-carrying capacity which could be linked with
the improvements in fitness [75] which may explain
the reported improvement in fitness measured by
CPET and the 6MWT. Therapeutic intermittent hypoxia
may be particularly useful in HFrEF patients not only
because HFrEF is associated with a reduced exercise
capacity but also because individuals with HFrEF have
a reduced ability to take up oxygen as well as impaired
peripheral oxygen utilisation [77].
Conclusion
The multifactorial aetiology of HF and its ancillary consequences is currently difficult to successfully manage with pharmacological interventions. Despite great advances and increases in the pharmacotherapeutic agents available for its management, HFrEF is a progressive condition and mortality remains high. TIH provokes adaptive changes to diverse pathways via altered metabolism and diminished cardiovascular risk factors and potentially benefit HF patients. These beneficial changes include but are not limited to: (i) decreased fatty acid synthesis, and circulating lipid levels with increased fatty acid metabolism; (ii) increased glucose uptake; (iii) decreased inflammatory markers; (iv) increased antioxidant defence; (v) decreasing sympathetic input via altered vagal tone; (vi) increased haemoglobin and red blood cell count, EPO and VEGF; and (vii) increased anti-inflammatory cytokines; and (viii) increased exercise tolerance.
The documented changes in both cardiovascular risk factors, exercise tolerance, circulating inflammatory markers together with the elevation of red blood cell number and haemoglobin levels pose potential benefits to be gained in heart failure patients. To date, there have been no trials in this area assessing whether TIH has similar results in the HF populace compared to other groups. Similarly, there is no assessment of the effects TIH on major adverse cardiovascular events or all-cause mortality in any population. Overall TIH presents a promising non-pharmacological modality that is worth exploring in the heart failure patient.
References
- Shah KS, Xu H, Matsouaka RA, Bhatt DL, Heidenreich PA, Hernandez AF, et al. Heart failure with preserved, borderline, and reduced ejection fraction: 5-year outcomes. J Am Coll Cardiol 2017;70(20):2476-2486.
- Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol 2016;13(6):368-378.
- Dharmarajan K, Rich MW. Epidemiology, pathophysiology, and prognosis of heart failure in older adults. Heart Fail Clin 2017;13(3):417-426.
- van Riet EE, Hoes AW, Wagenaar KP, Limburg A, Landman MA, Rutten FH. Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur J Heart Fail 2016;18(3):242-252.
- Zannad F, Ferreira JP, Pocock SJ, Anker SD, Butler J, Filippatos G, et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. The Lancet 2020;396(10254):819-829.
- Packer M, Anker SD, Butler J, Filippatos G, Ferreira JP, Pocock SJ, et al. Empagliflozin in patients with heart failure, reduced ejection fraction, and volume overload: EMPEROR-reduced trial. J Am Coll Cardiol 2021;77(11):1381-1392.
- Ilieșiu AM, Hodorogea AS. Treatment of heart failure with preserved ejection fraction. Adv Exp Med Biol 2018;1067:67-87.
- Philipson H, Ekman I, Forslund HB, Swedberg K, Schaufelberger M. Salt and fluid restriction is effective in patients with chronic heart failure. Eur J Heart Fail 2013;15(11):1304-1310.
- Völler H, Schwaab B. Cardiac rehabilitation. Der Kardiologe 2020;14:106-112.
- Costalat G, Lemaitre F, Tobin B, Renshaw G. Intermittent hypoxia revisited: a promising non-pharmaceutical strategy to reduce cardio-metabolic risk factors?. Sleep Breath 2018;22:267-271.
[Crossref][Google Scholar] [PubMed]
- Tobin B, Costalat G, Renshaw GM. Intermittent not continuous hypoxia provoked haematological adaptations in healthy seniors: hypoxic pattern may hold the key. Eur J Appl Physiol 2020;120(3):707-718.
[Crossref][Google Scholar] [PubMed]
- Gangwar A, Paul S, Ahmad Y, Bhargava K. Intermittent hypoxia modulates redox homeostasis, lipid metabolism associated inflammatory processes and redox post-translational modifications: benefits at high altitude. Sci Rep 2020;10(1): 7899.
[Crossref][Google Scholar] [PubMed]
- Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol 2014;76:39-56.
[Crossref][Google Scholar] [PubMed]
- Navarrete-Opazo A, Mitchell GS. Therapeutic potential of intermittent hypoxia: a matter of dose. Am J Physiol Regul Integr Comp Physiol 2014;307(10):R1181-97.
[Crossref][Google Scholar] [PubMed]
- Kimura H, Ota H, Kimura Y, Takasawa S. Effects of intermittent hypoxia on pulmonary vascular and systemic diseases. Int J Environ Res Public Health 2019;16(17):3101.
[Crossref][Google Scholar] [PubMed]
- Sarkar M, Niranjan N, Banyal PK. Mechanisms of hypoxemia. Lung India 2017;34(1): 47-60.
[Crossref][Google Scholar] [PubMed]
- Dempsey JA, Veasey SC, Morgan BJ, O'Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 2010;90(1):47-112.
- Michiels C. Physiological and pathological responses to hypoxia. Am J Pathol 2004;164(6):1875-1882.
[Crossref][Google Scholar] [PubMed]
- Briançon-Marjollet A, Weiszenstein M, Henri M, Thomas A, Godin-Ribuot D, Polak J. The impact of sleep disorders on glucose metabolism: endocrine and molecular mechanisms. Diabetol Metab Syndr 2015;7:25.
[Crossref][Google Scholar] [PubMed]
- Mackenzie R, Maxwell N, Castle P, Brickley G, Watt P. Acute hypoxia and exercise improve insulin sensitivity (SI2*) in individuals with type 2 diabetes. Diabetes Metab Res Rev 2011;27(1):94-101.
[Crossref][Google Scholar] [PubMed]
- Serebrovska TV, Grib ON, Portnichenko VI, Serebrovska ZO, Egorov E, Shatylo VB. Intermittent hypoxia/hyperoxia versus intermittent hypoxia/normoxia: comparative study in prediabetes. High Alt Med Biol 2019;20(4):383-391.
[Crossref][Google Scholar] [PubMed]
- Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol 2020;17(5):269-285.
[Crossref][Google Scholar] [PubMed]
- Medzhitov R. Origin and physiological roles of inflammation. Nature 2008;454(7203):428-435.
[Crossref][Google Scholar] [PubMed]
- Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, et al. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation 2003;108(1):79-85.
[Crossref][Google Scholar] [PubMed]
- Rahtu-Korpela L, Määttä J, Dimova EY, Hörkkö S, Gylling H, Walkinshaw G, et al. Hypoxia-inducible factor prolyl 4-hydroxylase-2 inhibition protects against development of atherosclerosis. Arterioscler Thromb Vasc Biol 2016;36(4):608-617.
[Crossref][Google Scholar] [PubMed]
- Wu N, Zhang X, Du S, Chen D, Che R. Upregulation of miR-335 ameliorates myocardial ischemia reperfusion injury via targeting hypoxia inducible factor 1-alpha subunit inhibitor. Am J Transl Res 2018;10(12): 4082-4094.
[Google Scholar] [PubMed]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986;74(5):1124-1136.
[Crossref][Google Scholar] [PubMed]
- Renshaw GM, Nikinmaa M. Oxygen sensors of the peripheral and central nervous system. Handbook of neurochemistry and molecular neurobiology. 2007;20:271-296.
- Tirpe AA, Gulei D, Ciortea SM, Crivii C, Berindan-Neagoe I. Hypoxia: overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int J Mol Sci 2019;20(24):6140.
[Crossref][Google Scholar] [PubMed]
- Nagao A, Kobayashi M, Koyasu S, Chow CC, Harada H. HIF-1-dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int J Mol Sci 2019;20(2):238.
[Crossref][Google Scholar] [PubMed]
- Arai T, Tanaka M, Goda N. HIF-1-dependent lipin1 induction prevents excessive lipid accumulation in choline-deficient diet-induced fatty liver. Sci Rep 2018;8(1):14230.
[Crossref][Google Scholar] [PubMed]
- Muangritdech N, Hamlin MJ, Sawanyawisuth K, Prajumwongs P, Saengjan W, Wonnabussapawich P, et al. Hypoxic training improves blood pressure, nitric oxide and hypoxia-inducible factor-1 alpha in hypertensive patients. Eur J Appl Physiol 2020;120:1815-1826.
[Crossref][Google Scholar] [PubMed]
- Azevedo Jr JL, Carey JO, Pories WJ, Morris PG, Dohm GL. Hypoxia stimulates glucose transport in insulin-resistant human skeletal muscle. Diabetes 1995;44(6):695-698.
[Crossref][Google Scholar] [PubMed]
- Thomas A, Belaidi E, Moulin S, Horman S, van der Zon GC, Viollet B, et al. Chronic intermittent hypoxia impairs insulin sensitivity but improves whole-body glucose tolerance by activating skeletal muscle AMPK. Diabetes 2017;66(12):2942-2951.
[Crossref][Google Scholar] [PubMed]
- Cartee GD, Douen AG, Ramlal TO, Klip A, Holloszy JO. Stimulation of glucose transport in skeletal muscle by hypoxia. J Appl Physiol 1991;70(4):1593-1600.
[Crossref][Google Scholar] [PubMed]
- Rodríguez C, Muñoz M, Contreras C, Prieto D. AMPK, metabolism, and vascular function. FEBS J 2021;288(12):3746-3771.
[Crossref][Google Scholar] [PubMed]
- Helkin A, Stein JJ, Lin S, Siddiqui S, Maier KG, Gahtan V. Dyslipidemia part 1—review of lipid metabolism and vascular cell physiology. Vasc Endovascular Surg 2016;50(2):107-118.
[Crossref][Google Scholar] [PubMed]
- Brooks GA, Butterfield GE, Wolfe RR, Groves BM, Mazzeo RS, Sutton JR, et al. Increased dependence on blood glucose after acclimatization to 4,300 m. J Appl Physiol 1991;70(2):919-927.
[Crossref][Google Scholar] [PubMed]
- Braun B, Mawson JT, Muza SR, Dominick SB, Brooks GA, Horning MA, et al. Women at altitude: carbohydrate utilization during exercise at 4,300 m. J Appl Physiol 2000;88(1):246-256.
[Crossref][Google Scholar] [PubMed]
- Naito R, Miyauchi K. Coronary artery disease and type 2 diabetes mellitus current treatment strategies and future perspective. Int Heart J 2017;58(4):475-480.
[Crossref][Google Scholar] [PubMed]
- Xia J, Yin C. Glucose variability and coronary artery disease. Heart Lung Circ 2019;28(4):553-559.
[Crossref][Google Scholar] [PubMed]
- Song K, Zhang Y, Ga Q, Bai Z, Ge RL. Increased insulin sensitivity by high-altitude hypoxia in mice with high-fat diet-induced obesity is associated with activated AMPK signaling and subsequently enhanced mitochondrial biogenesis in skeletal muscles. Obes Facts 2020;13(5):455-472.
[Crossref][Google Scholar] [PubMed]
- Wu S, Zou MH. AMPK, mitochondrial function, and cardiovascular disease. Int J Mol Sci 2020;21(14):4987.
[Crossref][Google Scholar] [PubMed]
- Wang Q, Liu S, Zhai A, Zhang B, Tian G. AMPK-mediated regulation of lipid metabolism by phosphorylation. Biol Pharm Bull 2018;41(7):985-993.
[Crossref][Google Scholar] [PubMed]
- Dengler F. Activation of AMPK under hypoxia: many roads leading to Rome. Int J Mol Sci 2020;21(7):2428.
[Crossref][Google Scholar] [PubMed]
- Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med 2016;48(7):e245.
[Crossref][Google Scholar] [PubMed]
- Sprick JD, Mallet RT, Przyklenk K, Rickards CA. Ischaemic and hypoxic conditioning: potential for protection of vital organs. Exp Physiol 2019;104(3):278-294.
[Crossref][Google Scholar] [PubMed]
- Hausenloy DJ, Yellon DM. Ischaemic conditioning and reperfusion injury. Nat Rev Cardiol 2016;13(4):193-209.
[Crossref][Google Scholar] [PubMed]
- Mallet RT, Manukhina EB, Ruelas SS, Caffrey JL, Downey HF. Cardioprotection by intermittent hypoxia conditioning: evidence, mechanisms, and therapeutic potential. Am J Physiol Heart Circ Physiol 2018;315(2):H216-H232.
[Crossref][Google Scholar] [PubMed]
- Przyklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P. Regional ischemic'preconditioning'protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993;87(3):893-899.
[Crossref][Google Scholar] [PubMed]
- Francula-Zaninovic S, Nola IA. Management of measurable variable cardiovascular disease'risk factors. Curr Cardiol Rev 2018;14(3):153-163.
[Crossref][Google Scholar] [PubMed]
- Steven S, Frenis K, Oelze M, Kalinovic S, Kuntic M, Bayo Jimenez MT, et al. Vascular inflammation and oxidative stress: major triggers for cardiovascular disease. Oxid Med Cell Longev 2019;2019:7092151.
[Crossref][Google Scholar] [PubMed]
- Golia E, Limongelli G, Natale F, Fimiani F, Maddaloni V, Pariggiano I, et al. Inflammation and cardiovascular disease: from pathogenesis to therapeutic target. Curr Atheroscler Rep 2014;16(9):435.
[Crossref][Google Scholar] [PubMed]
- Ivashkiv LB. The hypoxia–lactate axis tempers inflammation. Nat Rev Immunol 2020;20(2):85-86.
[Crossref][Google Scholar] [PubMed]
- D’Amario D, Cappetta D, Cappannoli L, Princi G, Migliaro S, Diana G, et al. Colchicine in ischemic heart disease: the good, the bad and the ugly. Clin Res Cardiol 2021;110:1531-1542.
[Crossref][Google Scholar] [PubMed]
- Leung YY, Hui LL, Kraus VB. Colchicine—update on mechanisms of action and therapeutic uses. InSeminars in arthritis and rheumatism 2015;45(3):341-350.
[Crossref][Google Scholar] [PubMed]
- Nidorf SM, Fiolet AT, Mosterd A, Eikelboom JW, Schut A, Opstal TS, et al. Colchicine in patients with chronic coronary disease. N Engl J Med 2020;383(19):1838-1847.
[Crossref][Google Scholar] [PubMed]
- Bouabdallaoui N, Tardif JC, Waters DD, Pinto FJ, Maggioni AP, Diaz R, et al. Time-to-treatment initiation of colchicine and cardiovascular outcomes after myocardial infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT). Eur Heart J 2020;41(42):4092-4099.
[Crossref][Google Scholar] [PubMed]
- Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci 2018;132(12):1243-1252.
[Crossref][Google Scholar] [PubMed]
- Murphy SP, Kakkar R, McCarthy CP, Januzzi Jr JL. Inflammation in heart failure: JACC state-of-the-art review. J Am Coll Cardiol 2020;75(11):1324-1340.
[Crossref][Google Scholar] [PubMed]
- Shirazi LF, Bissett J, Romeo F, Mehta JL. Role of inflammation in heart failure. Curr Atheroscler Rep 2017;19:1-9.
[Crossref][Google Scholar] [PubMed]
- Halade GV, Lee DH. Inflammation and resolution signaling in cardiac repair and heart failure. EBioMedicine 2022;79:103992.
[Crossref][Google Scholar] [PubMed]
- Verges S, Chacaroun S, Godin-Ribuot D, Baillieul S. Hypoxic conditioning as a new therapeutic modality. Front Pediatr 2015;3:58.
[Crossref][Google Scholar] [PubMed]
- Gangwar A, Sharma M, Singh K, Patyal A, Bhaumik G, Bhargava K, et al. Intermittent normobaric hypoxia facilitates high altitude acclimatization by curtailing hypoxia-induced inflammation and dyslipidemia. Pflugers Arch 2019;471:949-959.
[Crossref][Google Scholar] [PubMed]
- Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med 2011 Feb 17;364(7):656-665.
[Crossref][Google Scholar] [PubMed]
- Bärtsch P, Saltin B. General introduction to altitude adaptation and mountain sickness. Scand J Med Sci Sports 2008;18:1-10.
[Crossref][Google Scholar] [PubMed]
- Hill GW, Gillum TL, Lee BJ, Romano PA, Schall ZJ, Kuennen MR. Reduced inflammatory and phagocytotic responses following normobaric hypoxia exercise despite evidence supporting greater immune challenge. Appl Physiol Nutr Metab 2020;45(6):628-640.
[Crossref][Google Scholar] [PubMed]
- Anand IS, Gupta P. Anemia and iron deficiency in heart failure: current concepts and emerging therapies. Circulation 2018;138(1):80-98.
[Crossref][Google Scholar] [PubMed]
- Okonko DO, Mandal AK, Missouris CG, Poole-Wilson PA. Disordered iron homeostasis in chronic heart failure: prevalence, predictors, and relation to anemia, exercise capacity, and survival. J Am Coll Cardiol 2011;58(12):1241-1251.
[Crossref][Google Scholar] [PubMed]
- Kim TS, Hanak M, Trampont PC, Braciale TJ. Stress-associated erythropoiesis initiation is regulated by type 1 conventional dendritic cells. J Clin Invest 2015;125(10):3965-3980.
[Crossref][Google Scholar] [PubMed]
- Wojan F, Stray-Gundersen S, Nagel MJ, Lalande S. Short exposure to intermittent hypoxia increases erythropoietin levels in healthy individuals. J Appl Physiol 2021;130(6):1955-1960.
[Crossref][Google Scholar] [PubMed]
- Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol 2022;79(17):e263-e421.
- Naghshin J, Rodriguez RH, Davis EM, Romano LC, McGaffin KR, O'Donnell CP. Chronic intermittent hypoxia exposure improves left ventricular contractility in transgenic mice with heart failure. J Appl Physiol 2012;113(5):791-798.
[Crossref][Google Scholar] [PubMed]
- Jung WS, Kim SW, Park HY. Interval hypoxic training enhances athletic performance and does not adversely affect immune function in middle-and long-distance runners. Int J Environ Res Public Health 2020;17(6):1934.
[Crossref][Google Scholar] [PubMed]
- Burtscher M, Pachinger O, Ehrenbourg I, Mitterbauer G, Faulhaber M, Pühringer R, et al. Intermittent hypoxia increases exercise tolerance in elderly men with and without coronary artery disease. Int J Cardiol 2004;96(2):247-254.
[Crossref][Google Scholar] [PubMed]
- Albouaini K, Egred M, Alahmar A, Wright DJ. Cardiopulmonary exercise testing and its application. Postgrad Med J 2007;83(985):675-682.
[Crossref][Google Scholar] [PubMed]
- Guazzi M, Bandera F, Ozemek C, Systrom D, Arena R. Cardiopulmonary exercise testing: what is its value?. J Am Coll Cardiol 2017 Sep 26;70(13):1618-1636.
[Crossref][Google Scholar] [PubMed]
- Wicks JR, Turner GT, Leslie SL, Jayasinghe R. Changes Observed in the 6-minute Walk Test in Response to Exercise-based Cardiac Rehabilitation. Exerc Med 2022;6:2.
- Bayer U, Glazachev OS, Likar R, Burtscher M, Kofler W, Pinter G et al. Adaptation to intermittent hypoxia-hyperoxia improves cognitive performance and exercise tolerance in the elderly. Adv Gerontol 2017;7:214-220.
[Crossref][Google Scholar] [PubMed]
Copyright: © 2023 The Authors. This is an open access article under the terms of the Creative Commons Attribution NonCommercial ShareAlike 4.0 (https://creativecommons.org/licenses/by-nc-sa/4.0/). This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.